Running KPP¶
Now that KPP is properly compiled, we proceed to running the first test case to make sure it works! It’s advised to have the official KPP manual along with you during this section.
The first test case¶
We now follow the manual and begin running the Chapman stratospheric mechanism as a test case. This will allow us to illustrate some key features when running KPP.
In order to run a simulation on KPP, it needs three things:
- a
.kppfile (typels $KPP_HOME/examplesto see some examples of those) - a
.spcfile (typels $KPP_HOME/modelsto see some examples of those) - a
.eqnfile (typels $KPP_HOME/modelsto see some examples of those)
We begin by creating a directory to run this first test. Let’s call this
directory test1. We can create this directory anywhere: even inside KPP’s home
directory, although, for the sake of simplicity, let’s create it in your home directory:
cd $HOME
mkdir test1
Now let’s go to that directory with cd test1. Following the manual, let us
create a file called small_strato.kpp with the following contents:
#MODEL small_strato
#LANGUAGE Fortran90
#INTEGRATOR rosenbrock
#DRIVER general
You can do this by typing notepad++ small_strato.kpp in the test1
directory, if using Notepad++, or by using another editor of your choice
(replace notepad++ with gedit for example). Then just paste the content
above in the file, save and exit it.
This file tells KPP what model to use (small_strato.def) and how to process
it (most importantly for us here, it tells KPP to generate a Fortran 90 code,
although it can also generate C and Matlab code). Many other options can be
added to this file and you can learn more about them in the KPP manual.
If our changes to .bashrc are correct, then KPP should be able to find the
correct model, since the small_strato model (given by small_strato.def)
is located in the models directory, in the KPP home directory. We test this
by running KPP on our recently created file with
kpp small_strato.kpp
You should see the following lines on your screen:
This is KPP-2.2.3.
KPP is parsing the equation file.
KPP is computing Jacobian sparsity structure.
KPP is starting the code generation.
KPP is initializing the code generation.
KPP is generating the monitor data:
- small_strato_Monitor
KPP is generating the utility data:
- small_strato_Util
KPP is generating the global declarations:
- small_strato_Main
KPP is generating the ODE function:
- small_strato_Function
KPP is generating the ODE Jacobian:
- small_strato_Jacobian
- small_strato_JacobianSP
KPP is generating the linear algebra routines:
- small_strato_LinearAlgebra
KPP is generating the Hessian:
- small_strato_Hessian
- small_strato_HessianSP
KPP is generating the utility functions:
- small_strato_Util
KPP is generating the rate laws:
- small_strato_Rates
KPP is generating the parameters:
- small_strato_Parameters
KPP is generating the global data:
- small_strato_Global
KPP is generating the stoichiometric description files:
- small_strato_Stoichiom
- small_strato_StoichiomSP
KPP is generating the driver from none.f90:
- small_strato_Main
KPP is starting the code post-processing.
KPP has succesfully created the model "small_strato".
Note
If you get an error message here, go back a few steps and make sure the $KPP_HOME
and the $PATH variable are set correctly, and be sure that both KPP can be
found and the correct model files small_strato are in $KPP_HOME/models.
If indeed you see this output (or something very similar) it means you were
successful in creating the model. Now if you list your files with the ls
command, you’ll see many new files:
Makefile_small_strato small_strato.map
small_strato_Function.f90 small_strato_mex_Fun.f90
small_strato_Global.f90 small_strato_mex_Hessian.f90
small_strato_Hessian.f90 small_strato_mex_Jac_SP.f90
small_strato_HessianSP.f90 small_strato_Model.f90
small_strato_Initialize.f90 small_strato_Monitor.f90
small_strato_Integrator.f90 small_strato_Parameters.f90
small_strato_Jacobian.f90 small_strato_Precision.f90
small_strato_JacobianSP.f90 small_strato_Rates.f90
small_strato.kpp small_strato_Stoichiom.f90
small_strato_LinearAlgebra.f90 small_strato_StoichiomSP.f90
small_strato_Main.f90 small_strato_Util.f90
Most of them end with a .f90 extension, which tells us they are Fortran 90
codes. These codes have to be compiled into an executable file which is what
will actually process and run the kinetic model. So the next step is to compile
every one of those code together into one executable and run it. To do that,
let’s focus for now on the Makefile_small_strato. This is a text file that
tells your computer which Fortran compiler to use to compile, which files to
use, etc. We need to modify it, so open the Makefile_small_strato file
(again using your preferred editor) and find where it says
#COMPILER = G95
#COMPILER = LAHEY
COMPILER = INTEL
#COMPILER = PGF
#COMPILER = HPUX
#COMPILER = GFORTRAN
Each of the lines is a different Fortran compiler, and your computer is only
going to see the line that doesn’t start with a # (we say that the lines
with # are commented and therefore the computer doesn’t “see” them). So,
currently, these lines are telling the computer to use the Intel Fortran
compiler, ifort.
If you are using ifort, you should leave it as it is. However, ifort is
paid, so chances are you are using another compiler. If this is the case, put
the # in front of the INTEL options and take it out of the line which
has the name of your compiler. If you don’t know which compiler you have,
chances are you have gfortran, which is free and the one we will use here. You
can install gfortran with sudo apt install gfortran (or the equivalent
installation command for your system).
So, for gfortran, you should make the above lines of code look like
the following:
#COMPILER = G95
#COMPILER = LAHEY
#COMPILER = INTEL
#COMPILER = PGF
#COMPILER = HPUX
COMPILER = GFORTRAN
When doing that we say that we “uncommented” the gfortran line, since every
line that starts with a # is commented and not read by the system. You can
save and exit the file.
Now all you have to do is run the following command:
make -f Makefile_small_strato which will compile your Fortran code into an
executable file (.exe) using the options we just set. You should see a lot
of lines appearing on screen starting with gfortran, maybe some warnings,
and if no error messages appear the compilation was successful.
Now you’ll see many more new files, including one called small_strato.exe,
which is your executable file (run ls again list everything and see that).
This is the executable that will actually calculate the concentrations using
the model.
To test if it works, run the following command:
./small_strato.exe
which will run the executable. You should see some output on the screen with concentrations, like Fig. Output concentrations of the first test case.
Output concentrations of the first test case.
If this is the case, then your run was successful and everything worked well!
You just calculated the concentrations of the compounds in the small_strato
model with the pre-defined initial conditions.
Understanding the test case¶
Now let’s understand why our run of small_strato.exe was successful and what
happened. First, by running kpp small_strato, what we did was to tell KPP
to open a file called small_strato.kpp, in the current directory and do what that
file tells it to do. In the first line of the file there is the command
#MODEL small_strato
which tells KPP to look for a file called small_strato.def. Since the file
is in KPP’s models directory (at $HOME_KPP/models), KPP had no problems
finding it. This file has the initial concentrations you want to use in the
model, the time step, etc. It also links two other files (small_strato.spc
and small_strato.eqn), which tell KPP with chemical species and chemical
equations to use (effectively defining the mechanism).
After receiving all that information, KPP finally creates a Fortran 90 code
(because it says so in the small_strato.kpp we created) with our small
stratospheric model containing our pre-defined initial conditions, time step,
chemical reactions and so on.
The code, however, has to be compiled before run, so that is why we issued the
command make, which compiles the code according to the file
Makefile_small_strato (which is where we specified the Fortran compiler).
This step creates an executable file, which has the extension .exe and is
ready to be run. By running the .exe file we ran a program that got our
initial concentrations of the species we defined and, based on the chemical
reactions, calculated, step by step, their concentrations in each time step.
At each step, the model is not only printing the concentrations on screen, but
it is also writing them into a file called small_strato.dat, which is a
column-separated text file. This file can be used to see, plot, make
calculations with the data and so on. However, you should be careful because
the order of the concentrations that appear on screen isn’t the same order KPP
uses for the .dat file. You can learn about the ordering at page 7 of the
KPP manual, but a good rule of thumb is to check the file with a .map
extension (in this case, small_strato.map) and take a look at the
species section. The file output order is the ordering of the variable
species followed by the species on the fixed species.
In the case of small_strato the order printed on the file (you can check it on
small_strato.map) is
time, O1D, O, O3, NO, NO2, M, O2
The time is always going to be the first column, and it is always going to be
in hours since the start of the simulation. Since the solar forcing matters
here, we need to keep track of the time of day that the simulation started. In
this case it was at noon, because that’s the way the .def file is set (we
will talk about this in more detail in the sections to come).
We can read that data in many ways. I present below a quick python script to plot the concentrations as a function of the hour of the day
1 2 3 4 5 6 7 | import pandas as pd
from matplotlib import pyplot as plt
concs = pd.read_csv('small_strato.dat', index_col=0, delim_whitespace=True, header=None).apply(pd.to_numeric, errors='coerce')
concs.columns = ['O1D', 'O', 'O3', 'NO', 'NO2', 'M', 'O2']
concs.index.name = 'Hours since noon'
concs.plot(ylim=[1.e8, None], logy=True, y=['O3', 'NO', 'NO2'], grid=True)
plt.savefig('test1_time.png')
|
Note
KPP has a small issue with formatting and sometimes prints a number that can’t
be read because some strings are missing. For example, printing 3.4562-313.
This can’t be normally read and it’s supposed to be 3.4562E-313 and this
(apparently) only happens when the number is close to machine-precision (which
we would interpret as zero). The program above takes this issue into
consideration (in line 3) when reading the file, but you should pay attention to that when
trying to read with by other means.
If you have ever seen python before, this code should be pretty intuitive. If
you haven’t you can still use it easily (depending on how you got python, you
might have to install python’s pandas package). This code generates the
following plot of the concentrations:
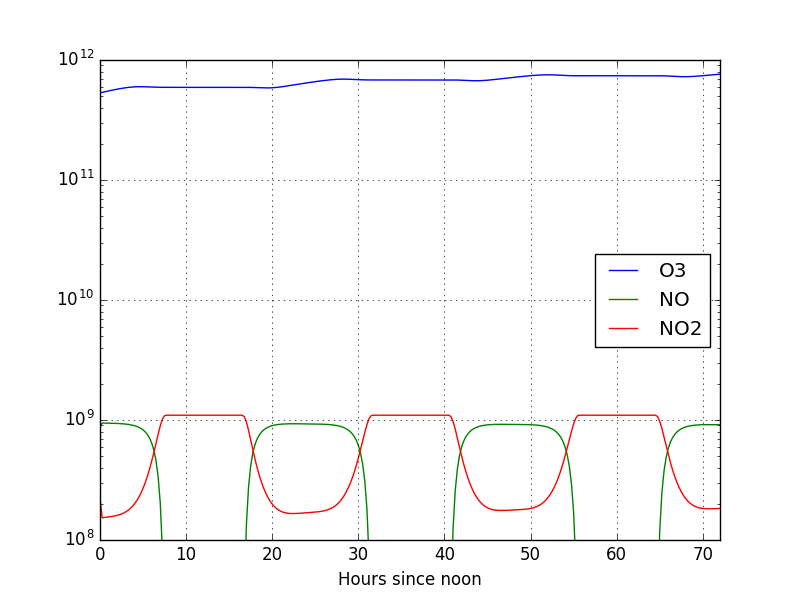
We can see that the NOx concentrations follow the solar cycle, which is
indicative that the model is indeed working properly. However we see that the
O3 concentrations still haven’t stabilized. This tells us that we need to run
the model for longer. Let us take this chance to modify the small_strato
example a bit, try and make the O3 concentrations stabilize and learn how to
alter/create models.
